

Wei Chi instrument
Your laboratory is always a good helper
目前,肿瘤免疫治疗方兴未艾,群雄并起,其中有效的治疗策略之一就是过继细胞转移疗法(ACT)。嵌合抗原受体(CAR)和工程化T细胞受体(TCR)是近年来主要的过继性T细胞免疫疗法。TCR工程T细胞表达肿瘤抗原特异性受体,其α链和β链由高质量、高亲和力的抗原特异性T细胞克隆产生。
TCR分子属于免疫球蛋白的一个超家族,由两个共价结合的多态性亚单位组成,每个亚单位都是抗原特异性的,它们至少与四种不同类型的信号转导链有关。为了激活T细胞,TCR和主要组织相容性复合体(MHC)之间必须存在相互作用。
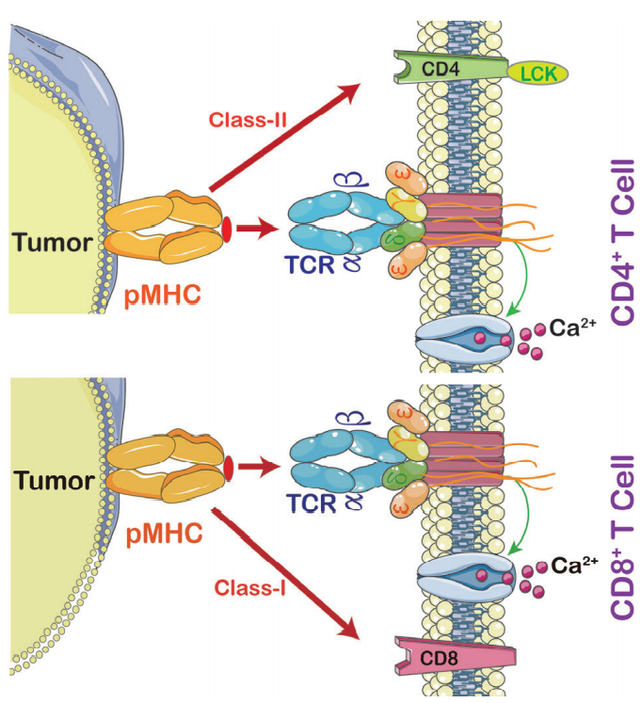
TCRs与pMHC(peptide-MHC)相互作用的强弱决定了未成熟胸腺细胞的命运,对幼稚T细胞的存活至关重要。因此,TCR-T免疫治疗技术通过与MHC特别是Ⅱ类分子的有效相互作用激活宿主的免疫系统,后者被TCR-T细胞和CAR-T细胞特异识别。TCR-T细胞可以识别细胞内的肿瘤特异性抗原,而CAR-T细胞主要识别肿瘤表面的特异性抗原。这使得TCR-T细胞在肿瘤治疗中更有效。
目前,一些新的技术和工具正在应用于TCR-T,有助于提高TCR-T治疗的疗效和安全性,TCR-T细胞治疗正展现出抗肿瘤治疗的巨大潜力。
CAR-T与TCR-T的比较
在ACT疗法中、TCR-T和CAR-T细胞都已被成功地用于实体瘤的临床治疗。
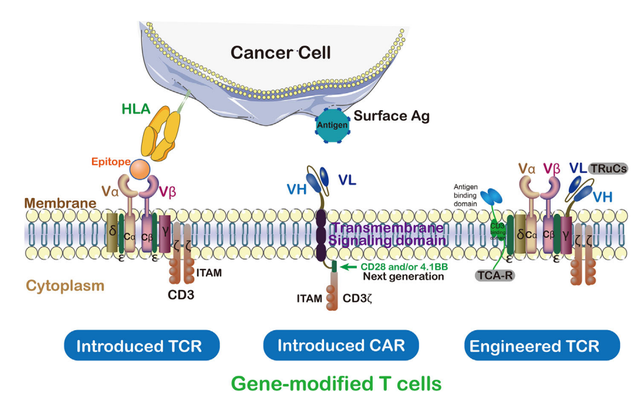
CAR包含肿瘤抗原靶向的单链抗体、跨膜结构域和CD3ζ的胞内激活结构域。通过这种方式,工程化的CAR能够识别特定的肿瘤相关抗原,CAR能够在不经过MHC处理的情况下结合未经处理的肿瘤表面抗原。
代CAR-T细胞表现出有限的扩增和相对较短的持久性, “第二代”CAR-T加入了共刺激受体CD28、4-1BB/CD137和OX40。将这些共刺激受体添加到CAR-T细胞的CD3ζ结构域中,从而促进更强大和持久的T细胞反应。第三代CARs同时结合两个共刺激信号(CD28和4-1BB),这比第二代CARs具有更好的扩增和更长的持续性。
相反,TCR是与MHC抗原复合物结合的α/β异二聚体。与TCR相比,CARs识别肿瘤抗原具有某些缺点,如肿瘤外毒性。与CARs相比,TCRs在基于T细胞的治疗中具有一些结构性优势,例如其受体结构中有更多的亚单位(10:1),免疫受体基于酪氨酸的激活基序(ITAMs)更多(10:3),对抗原的依赖性更小(1:100),以及更多的共刺激受体(CD3,CD4,CD28等等)。具有低MHC亲和力范围(104-106M-1)的TCR就可以有效的激活T细胞,相反,CARs需要更高的亲和力范围(106-109M-1)。
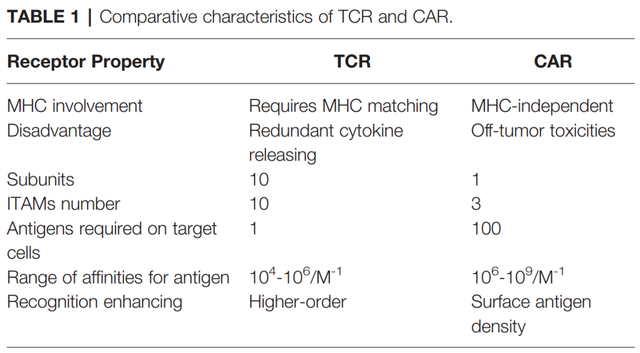
因此,CAR介导的细胞毒性依赖于更高密度的细胞表面抗原。此外,T细胞/抗原相互作用在免疫突触(IS)结构中启动,其中TCR呈现具有外周LFA-1粘附的环状区域,而CAR呈现无环状区域的弥散LFA-1分布。因此,TCR-IS比CAR-IS发出的信令速度慢但持续时间长。同时,CAR-T细胞呈现出更快的杀伤功能,并向下一个肿瘤靶点迁移(连环杀伤),这与TCR-T细胞延长信号传导和延长杀伤时间形成鲜明对比。
重组TCRs
TCR是人体复杂的受体之一,它包含六种不同的受体亚单位,它们在T细胞中具有非常广泛的功能。肿瘤浸润淋巴细胞(TILs)的TCR的改变显著影响肿瘤特异性T细胞。其中TCR的变化有助于T细胞的增殖,TCR多样性与抗肿瘤作用相关。
对TIL的TCR工程化是肿瘤的佳治疗方法之一。TCR由结合到肽-MHC配体的α链和β链、CD3复合体的信号亚单位(ϵ、γ和δ)以及CD3ζ同源二聚体组成。除CD3ζ外,所有亚单位均具有细胞外免疫球蛋白(Ig)结构域。基于这些结构,利用工程化TCR的新技术有ImmTAC、TRuCs和TAC等。
免疫动员单克隆T细胞受体(ImmTAC)
ImmTACs是使用工程化、可溶性和亲和增强的单克隆TCRs(mTCRs)设计的。ImmTACs基本上是融合蛋白,结合了工程化的TCR靶向系统和单链抗体片段(scFv)效应器功能。在ImmTACs的构建中, TCR能够识别来自人类白细胞抗原(HLA)呈递的细胞内靶点的肽。
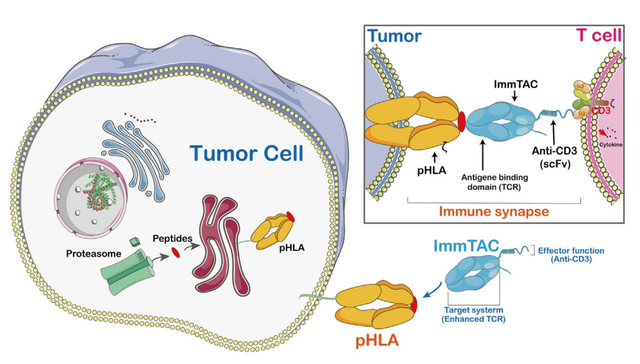
ImmTAC通过特异性靶向肿瘤细胞表面的HLA-肽复合物,并通过scFv抗体片段与CD3的相互作用促进T细胞介导的效应器功能。ImmTAC还以剂量依赖性方式激活CD8+T细胞,并能有效地重定向和激活效应和记忆CD8+和CD4+细胞。ImmTAC通过分泌多种细胞因子表现出多功能反应,如TNF-α、IFN-γ、IL-6、MIP1α-β和IFN-γ诱导蛋白10。
此外,选择合适的靶抗原是ImmTACs的关键,质谱技术和MHC多聚体技术有助于识别合适的抗原。值得注意的是,TCR工程化T细胞也表现出非预期靶向毒性。总的来说,ImmTACs已被证明能增强TCR-T细胞的抗肿瘤反应,但其安全性有待进一步研究。
T细胞受体融合结构
T细胞受体融合结构(TRuCs),一种与T细胞受体亚单位融合的抗体结合域,设计用于有效识别肿瘤表面抗原。TRuCs由靶向肿瘤相关抗原的特异性抗体融合到5个TCR亚基(TCRα、TCRβ、CD3ϵ、CD3γ和CD3δ)的胞外N-末端组成,为工程化T细胞提供了新的靶向特异性和HLA非依赖性靶细胞清除能力,可被相应的靶细胞激活。
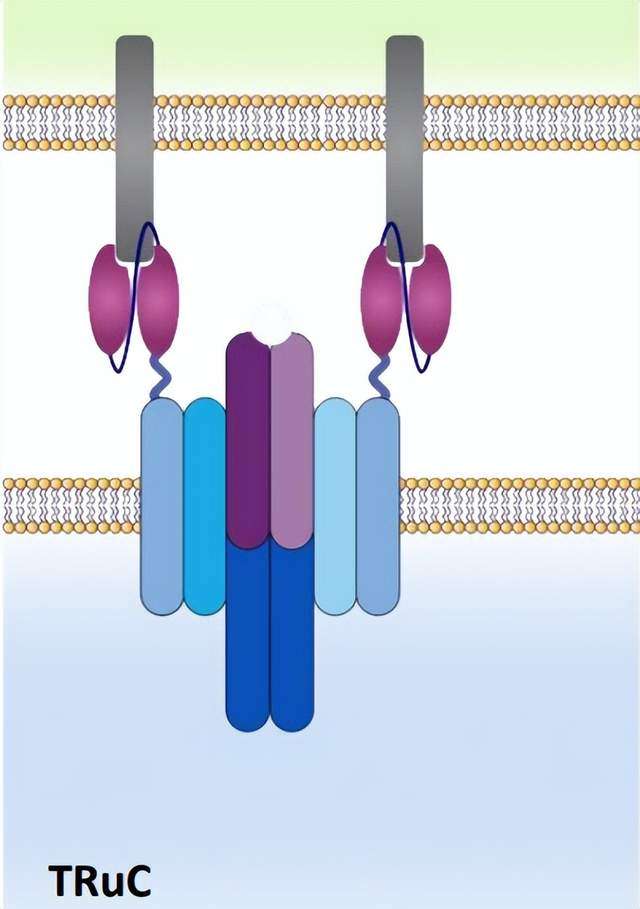
与第二代CAR-T细胞相比,该方法显示出更好的抗肿瘤效果。此外,TRuCs支配TCR复合体的全部信号机制,而CARs仅利用分离的CD3ζ胞内段的有限信号。
T细胞抗原偶联剂(TAC)
T细胞抗原偶联剂是另一个工程化TCR细胞,以非MHC依赖性方式诱导更有效的抗肿瘤反应并降低毒性。TAC嵌合蛋白通过与CD3结构域的结合,从而形成TCR/CD3复合物并获得更多的T细胞应答。
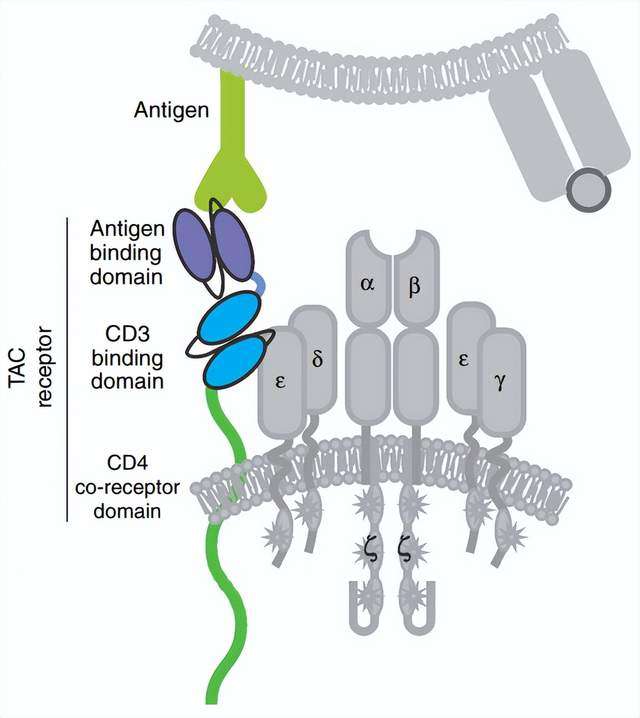
TAC受体的活性与CD3结合域的选择密切相关。例如,与UCHT1相比,来自OKT3(muromonab-CD3)的单链抗体具有较低的细胞因子产生和细胞毒性,这可能导致实质上不同的功能结果。与第二代CARs相比,TAC基因工程化的T细胞不仅有利于过继后在实体瘤的更大浸润,而且减少了T细胞在表达抗原的健康组织中的扩增和肿瘤外毒性。
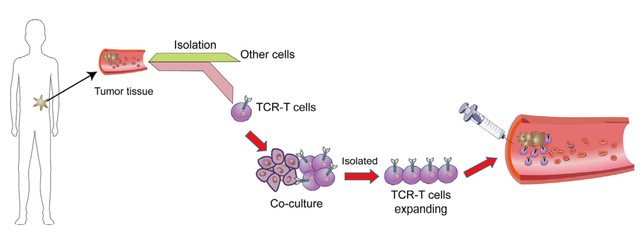
TCR-T细胞疗法的工作流程
为了分离治疗性TCR,必须首先从患者或健康献血者的血液中分离抗原特异性T细胞,并在体外用特异性肽抗原以及细胞因子(如IL-2和IL-15)进行扩增。这一过程需要事先确定可安全靶向于患者的特定肿瘤相关肽靶点。在选择了靶抗原后,可以使用不同的方法来筛选具有所需的高亲和力和肿瘤特异性的TCR。临床前安全性测试对于确保分离的高亲和力TCR的小靶外效应和交叉反应性也是必要的。病毒载体通常用于对自体患者T细胞进行基因修饰,以表达经验证的治疗性TCR,然后再输回患者体内。
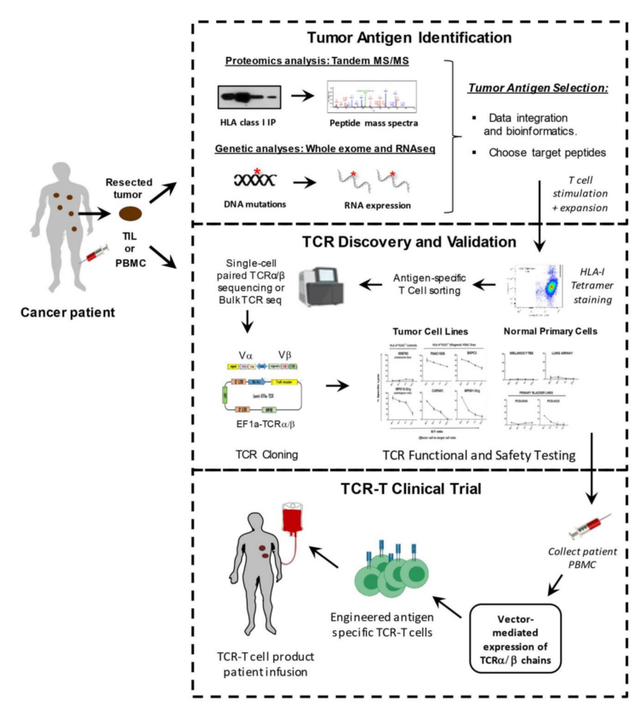
确定靶抗原
T细胞识别的黑色素瘤抗原1(MART-1)是TCR-T临床试验中个靶向的肿瘤相关抗原。在这一突破之后,针对多种肿瘤抗原的TCR-T疗法已经开发出来,包括针对MAGE-A3、MAGE-A4、GD2、间皮素、gp100、MART1、AFP、CEA、NY-ESO-1以及源自HPV和EBV的病毒肽的TCR-T疗法。其中,NY-ESO-1已被证明是TCR-T细胞有希望的靶点之一,在治疗滑膜肉瘤方面取得了成功,客观有效率为67%。
理想的TCR-T靶抗原显示以下特征:(1)诱导免疫反应的能力;(2)与驱动肿瘤表型(如癌基因)相关,以降低抗原丢失和肿瘤免疫逃避的风险;以及(3)在肿瘤干细胞上的表达以促进永久性肿瘤根除。
肿瘤相关抗原的鉴定方法
高分辨率质谱(MS)已被证明是促进从肿瘤细胞直接识别HLA-I结合肽的强大的高通量方法。在这种方法中,通过免疫沉淀(IP)从肿瘤组织或细胞系中分离HLA-I/肽复合物,然后充分洗涤并应用酸性洗脱缓冲液,从HLA-I分子和用于IP的抗体中分离结合肽抗原。该策略允许每个肿瘤样本识别数千个经验证的肽靶点,并已用于识别胶质母细胞瘤(GB)、黑色素瘤、肾细胞癌(RCC)和结直肠癌(CRC)等的HLA-I配体。
肿瘤新抗原的鉴定方法
尽管基于MS的技术可用于识别新抗原,但由于其相对较低的丰度以及MS的有限灵敏度,尤其是对于大小有限的肿瘤样本,它们更难识别。然而,新一代测序技术的发展有助于识别和定位这类肿瘤靶点。全外显子组DNA测序,结合计算预测算法,允许识别癌细胞中的特定遗传改变,这些改变可以产生突变肽,并能够呈现在肿瘤HLA-I分子上。
所有体细胞突变基因都可以进行电脑分析,以预测可能与患者个体HLA-I分子结合的潜在高亲和力表位,从而被T细胞识别。随着大型MS洗脱肽数据库的使用,HLA-I肽结合预测算法不断更新和改进,其他预测算法试图考虑与细胞内过程复杂性相关的生物变量。
另一个经常使用的方法是肿瘤RNA测序,它允许选择具有高转录表达的新抗原。值得注意的是,尽管这些预测方法在识别呈递的和高免疫原性新抗原时通常表现出非常好的准确性,但它们通常预测的新抗原靶点的数量比真实靶点的实际数量高1到2个数量级。
通过trogocytosis发现新抗原是近年来出现的一种新方法。Trogocytosis是细胞结合过程中发生的一种生物学现象,在此过程中,细胞共享并转移膜和膜相关蛋白。Li等人发现T细胞膜蛋白特异性转移到肿瘤靶细胞,这些靶细胞呈递同源HLA-I/肽复合物。利用这些T细胞-靶细胞相互作用,他们通过将表达标记孤儿TCR的T细胞与同源靶细胞共同孵育,创建了新抗原发现系统。通过将荧光标记从T细胞转移到靶细胞,该方法能够分离这些靶细胞并对同源TCR配体进行测序,从而建立新抗原文库。
分离肿瘤特异性T细胞和TCR
使用HLA-I多聚体、单细胞TCR测序或抗原阴性的人源化小鼠,肿瘤反应性T细胞和TCR可从自体、异体或异种细胞库中识别鉴定。
利用HLA-I多聚体法,可通过多聚体染色和流式细胞术分选直接分离抗原特异性CD8+T细胞。在分离成对的全长TCR序列之前,对这些多克隆T细胞进行同源肽识别和抗肿瘤功能测试。采用高灵敏度的基于PCR的单细胞TCR分析方法(TCR-SCAN),可以得到具有高亲和力和特异性的TCR。
另外一种方法利用了人源化小鼠TCR基因库,该基因库不会发生在人类中产生的T细胞克隆缺失或耐受。为此,Li等人利用整个人类TCRα/β基因位点和嵌合HLA-A2转基因构建了转基因小鼠,以实现针对人类TAA的人类TCR的分离。
单细胞测序方法代表了高通量分离肿瘤特异性TCR编码基因的更有前景的方法。使用靶向TCRα和TCRβ基因座内每个单独V和J元件的RNA诱饵文库,可以从剪切的基因组DNA(gDNA)片段中选择性分离TCR编码基因组元件,用于随后的配对末端深度测序。这使得从人类材料或TCR人源化小鼠的寡克隆T细胞群中鉴定抗原特异性TCR成为可能。
幼稚T细胞也可以作为TCR-T治疗的TCR来源。TAA和新抗原特异性T细胞可从癌症患者外周血中的低频前体中衍生和扩增,并可重新输注或用作抗原特异性TCR的来源。由于癌症患者通常表现出免疫抑制或显性T细胞耐受,HLA-I匹配的健康供体的原始序列也代表了一个可靠的来源,因为它具有巨大的多样性TCR序列,理论上T细胞具有任何抗原特异性,包括肿瘤新抗原。已经开发了高通量技术平台,用于寻找原始的序列,以便快速有效地识别用于个性化过继性T细胞治疗的罕见但有治疗价值的TCR。
TCR的克隆
大多数基于TCR的基因治疗方法依赖于用病毒载体对T细胞进行体外转导,早用于基因治疗的载体是腺病毒。然而,由于它们不能将转基因整合到宿主基因组中,TCR表达在T细胞增殖过程中会丢失。此外,腺病毒的免疫遗传特性也限制了其作为基因治疗载体的应用。相比之下,逆转录病毒作为基因转移载体表现出更大的前景,因为它们可以感染多种细胞,并有能力将转基因插入宿主基因组,从而使异位TCRα/β链稳定表达。
由γ-逆转录病毒如小鼠白血病病毒(MLV)衍生的逆转录病毒载体已被广泛用于基因转移到人类T细胞中。这种方法已被用于传递各种基因,包括自杀基因、TCR和CARs。主要缺点是它们不能转导非增殖性靶细胞,这就排除了静止的T细胞在TCR-T治疗中的应用。此外,逆转录病毒插入突变可能引起潜在的副作用。
近,慢病毒载体(LV)作为基因转移载体获得了更多的关注,因为它们可以将基因传递到分裂和非分裂细胞中。各种技术,如Golden Gate克隆和LR克隆,通常用于构建插入TCRα/β基因的载体。
腺相关病毒(AAV)是另一种广泛使用的病毒载体。与腺病毒载体相比,AAV具有较低的免疫原性和更广泛的细胞向性,因此在肿瘤基因治疗中得到了广泛的应用。为了促进转基因整合,人们开发了自互补AAV载体(scAAV),使AAV独立于宿主细胞的互补链合成,scAAV在临床前模型中的疗效优于传统AAV。
同时,一些非病毒基因编辑方法也被开发出来。mRNA电穿孔已被证明可实现短暂的TCR和CAR表达,从而将病毒成分持续存在的风险降至低。临床数据表明,mRNA修饰的TCR-T和CAR T细胞都是可行和安全的,没有明显的证据表明对正常组织有非靶向毒性。然而,缺乏持续的TCR表达可能会限制疗效,需要反复输注。此外,非病毒性睡美人反转录转座子系统也被用于TCR和CARs的转导。
基因编辑通过同源定向修复(HDR)可以将大基因片段特异、高效地插入靶细胞。使用CRISPR/CAS9开发的TCR-T细胞已被证明在体外特异性识别肿瘤抗原,并在体内诱导产生性抗肿瘤反应。
TCR的验证方法
在TCR克隆后,需要进行广泛的临床前验证,以证明工程化TCR-T细胞的特异性和安全性。验证包括通过滴定同源肽抗原来评估TCR的亲和力,以及测量一组对HLA-I匹配的肿瘤细胞系的杀伤作用。如果没有这样的肿瘤细胞系存在,靶细胞可以转导表达相关抗原和相关的HLA-I分子。新抗原也可以在自体抗原呈递细胞中表达,以评估TCR的抗原反应性。
安全性测试包括测试候选TCR-T对HLA-I匹配原发组织的识别能力,以确保没有正常组织作为靶点,产生可能导致的非靶向毒性。在至少两次TCR-T细胞治疗临床试验中,发生过对正常脑细胞和心脏细胞的交叉反应,从而导致患者死亡。这些试验结果强调了TCR进入临床试验前进行广泛安全性试验的重要性。
TCR-T的安全性
TCR-T细胞的ACT显示出很高的肿瘤杀伤,但在一些临床研究中也出现了一些严重的不良事件。优化工程化T细胞中的TCR亲和力至关重要,受体亲和力能够决定T细胞治疗的安全性和有效性。就疗效而言,亲和力TCR相互作用足以激活T细胞,但需要强亲和力来维持T细胞的扩增。
在I/II期ACT临床试验中,低亲和力工程化T细胞显示出更安全的特性,但它们的抗肿瘤反应较弱。通过识别T细胞的TCR-pMHC相互作用,可以将工程化T细胞分为高亲和力型和低亲和力型。此外,人们也开发了一些技术来提高TCR-T的安全性。
基于工程化T细胞的安全开关机制是一种有吸引力的策略。来源于单纯疱疹病毒Ⅰ型(HSV-TK)的胸苷激酶基因是常见的自杀基因之一。
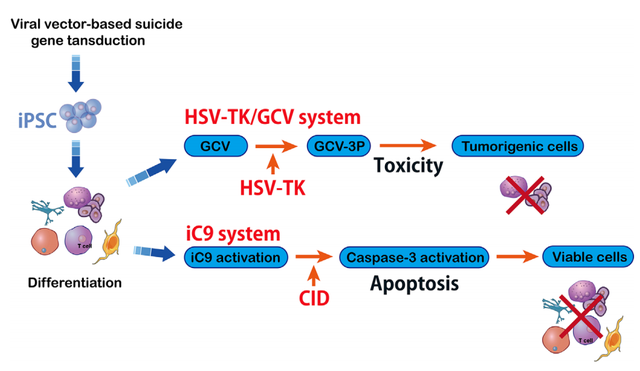
尽管HSV-TK在基于细胞的免疫治疗中显示出安全性,但需要导入磷酸化的核苷类似物。另一个更安全的诱导T细胞安全开关称为诱导型caspase-9(iC9)。iC9是一种修饰的人FK结合蛋白,可通过小分子化合物AP1903激活,这一过程依赖于线粒体凋亡途径。
iC9自杀基因的免疫原性较低,引发针对转基因细胞的免疫反应降低。基于iC9的安全开关已经被证明比先前的自杀基因更有潜力用于细胞治疗。
TCR-T细胞治疗的临床现状
截止2021年8月9日,在ClinicalTrials上共有175项使用TCR-T疗法的研究正在进行中,其中71项是针对特定TAA或新抗原的特异性TCR,有32项研究已经完成。NY-ESO-1是常见的靶向抗原,在多种癌症中均有表达,包括骨髓瘤、黑色素瘤等。其他肿瘤睾丸相关抗原,如PRAME和MAGE蛋白,以及黑色素瘤分化抗原MART-1和gp100,以及近的癌症驱动因子,如WT1、KRAS和TP53,也是流行的TCR-T靶点。
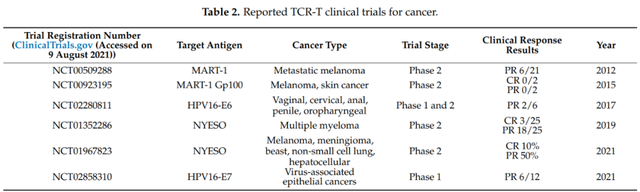
共有83名赞助者/合作者发起或参与TCR-T细胞治疗的研究,包括美国国家卫生研究院(NIH)、政府组织、行业和大学/学术机构。目前,美国国家癌症研究所(NCI)共支持了53个TCR-T项目,占到了所有正在进行项目的20%。
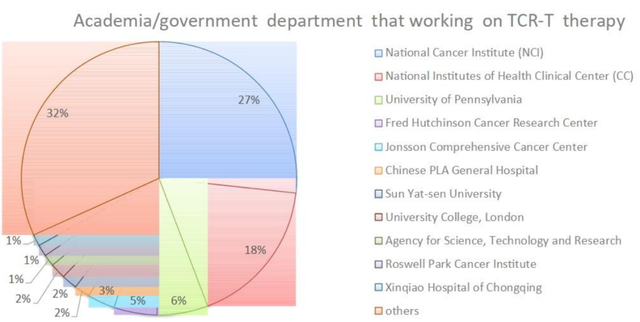
在开发TCR-T疗法的29家制药公司中,葛兰素史克和Adaptimunime发起了多的临床试验,分别为11项和7项。近,报道了一项针对人乳头瘤病毒(HPV)-16 E7蛋白的TCR-T细胞治疗转移性人乳头瘤病毒相关上皮癌的1期临床试验(NCT02858310)。在这项研究中,12名接受治疗的患者中有6名出现了客观的临床反应,观察到了稳健的肿瘤消退。这是TCR-T细胞疗法的一个里程碑式的临床试验,证明靶向病毒抗原对病毒相关癌症患者具有良好的临床效果。其他被探索为TCR靶点的病毒抗原包括HPV-E6蛋白、来自EB病毒(EBV)的抗原和人类内源性逆转录病毒(HERV)靶点,如HERV-E。
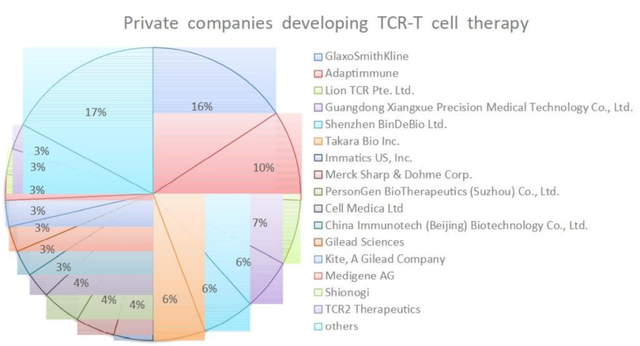
靶向TAAs 的MART-1和NY-ESO-1 TCR-T疗法在晚期黑色素瘤、骨髓瘤和非小细胞肺癌中也显示出临床疗效。完成的TCR-T临床试验的总有效率(ORR)在0~60%之间。值得注意的是,这些TCR-T临床试验中的大多数都只入组了少量患者(2至25名),因此ORR在统计上可能不太准确。因此,需要更大规模的II期和III期临床试验来确认这些TCR-T疗法的实际临床疗效。
TCR-T细胞治疗的挑战和潜在解决方案
尽管基于TCR-T细胞的免疫疗法已在大部分接受治疗的患者中显示出一定的临床疗效,但在许多领域仍然面临着诸多挑战。这些挑战包括:(1)靶向正常组织引起的免疫毒性;(2)工程化T细胞中TCR表达不足或短暂表达;(3)T细胞耗竭和功能障碍;(4)肿瘤免疫逃逸,以及(5)大多数癌症患者缺乏有效的肿瘤特异性抗原作为靶点。克服这些挑战将是未来取得更大临床成功的关键。
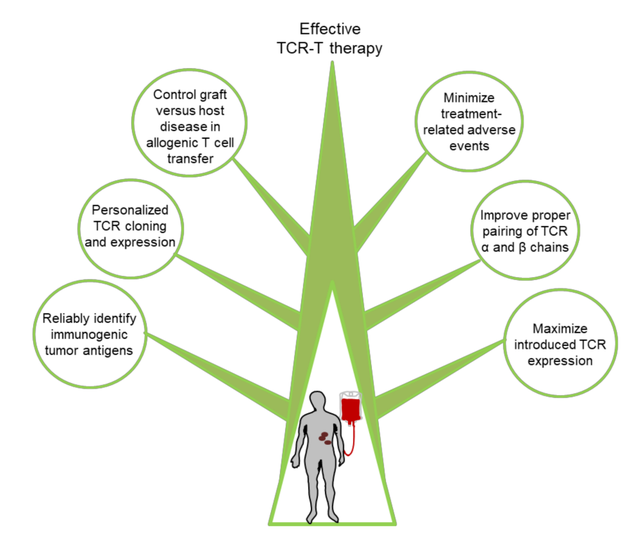
新靶标的发现
目前,用于TCR-T的有效和安全免疫治疗的肽抗原靶点非常有限。目前使用的大多数靶点是TAA,尽管在肿瘤组织中上调,但在正常组织中仍保持低水平的表达,这可能导致自身免疫毒性。因此,新抗原似乎是TCR-T癌症治疗安全的靶点。然而,在TCR-T临床开发新抗原的主要挑战包括:(1)新抗原形成突变在很大程度上是个体化的,并且在癌症患者之间存在差异,因此难以开发出广泛应用的免疫治疗产品;(2)新抗原在肿瘤组织中的表达常常是异质性的。
尽管如此,近年来的报告强调了肿瘤细胞广泛共享的免疫原性新抗原的出现,包括突变的KRAS和TP53。许多其他研究也证明了可用于产生潜在治疗性肿瘤特异性TCR的共享新抗原的免疫原性。随着下一代测序技术的发展,特别是单细胞DNA测序、转录组测序和成熟的体外验证方法,以个性化新抗原为靶点的TCR-T免疫治疗可能在未来几年成为一种流行的癌症治疗方法。此外,新出现的TAA类别,如癌胚抗原,也可能构成未来TCR-T发展的可行靶标。
大化治疗性TCR表达
转基因α和β链的正确配对是阻碍TCR-T细胞发展的主要挑战之一。由于每个转导的T细胞包括两条内源性TCR链和两条转化的TCR链,因此具有未知特异性的异二聚体可导致潜在的自身免疫后果。另一个相关问题是,不恰当的α/β链TCR配对将竞争CD3复合物,从而降低治疗性TCR的表面表达和信号转导。
有几种方法可对转导的TCR链进行适当配对,包括:(1)部分鼠源化TCR的恒定区;(2)添加半胱氨酸残基以促进引入TCR链的二硫键;(3)改变内源性TCR恒定区的二级结构;(4)向转导TCR的细胞内部分添加信号域;(5)将TCR-α/β链引入替代效应细胞或构建单链TCR。
增强治疗性TCR表达的方法包括:(1)TCR-α和TCR-β链转基因的密码子优化,以及(2)改变TCR-α/TCR-β载体配置以优化表达。
减少不良事件
通常,靶向非肿瘤毒性是TAA的主要关键障碍,这种风险促使研究人员更仔细地研究共同的新抗原。目前,多个癌基因热点突变正在被研究作为潜在的TCR靶点,如磷酸肌醇-3-激酶(PI3K)、KRAS和TP53。此外,通过带有自杀基因的基因工程化的TCR-T细胞是采用的一项重要安全措施。显然,发展可靠识别的个性化、高度特异性和免疫原性的肿瘤抗原靶点对于减少与TCR-T细胞治疗相关的不良事件至关重要。
异体T细胞的移植物抗宿主病
使用同种异体T细胞是一个非常有希望的方案,可以克服制造问题、患者相关免疫细胞缺陷和治疗延迟。为了使用同种异体T细胞,有必要控制由转导的同种反应性淋巴细胞引起的移植物抗宿主病以及宿主免疫系统对工程化淋巴细胞的排斥。
内源性TCR基因、HLA-I位点或CD52分子的缺失是避免TCR-T移植失败的策略之一,可通过多种方法实现,如基因编辑或使用siRNA。此外,多能干细胞技术也被认为是一种潜在的解决方案。
小结
近年来,工程化T细胞在治疗血液瘤方面显示出为优越的疗效。TCR调节对于T细胞的再活化、免疫应答及其对外来抗原的临床效应至关重要。而TCR-T细胞具有CAR-T无法比拟的优势,在临床前和临床研究中显示出巨大的潜力。
然而,提高TCR-T免疫治疗的抗肿瘤疗效仍然有几个关键挑战,包括如何安全地增加治疗性TCR的亲和力,如何在患者群体中鉴定共有的肿瘤特异性抗原和TCR,以及如何调节TCR的表达并实现佳功能。这些问题的解决将有助于充分发挥TCR-T细胞治疗的潜力,给肿瘤患者解除病痛带来希望。
文章来源:小药说药
重组C因子与基于LAL的测试方法具有可比性。使用PyroGene 重组C因子产品进行的4种产品内毒素检测时,结果均在可接受范围以内。对于具体产品的方法选择,可以根据产品的性质来进行确定。
赛桥生物致力于实现CGT工业级核心装备的封闭化、自动化、柔性化、 数字化和智能化,加速推动国产替代,已构建设备与耗材能力平台,完成全套工艺模块的开发,包括全封闭自动化血细胞分离、磁珠激活分选、电转染、细胞扩增、清洗浓缩、制剂分装等设备,以及配套的系列化GMP一次性密闭耗材。力争突破进口垄断、为行业客户提供完全国产自主可控的CGT数字化工艺装备平台。
低温样品的移动基站——斯特林 Stirling ULT25NEU 车载便携式超低温冰箱可用于远程临床试验样本采集、实验室小批量样本存储和生物药物配送,为实验室、科研机构生物样品的超低温贮存和运输赋能。
Shanghai Weichi Instrument Co.,
(Shanghai·Suzhou· Nanjing·Hangzhou)
2nd Floor, Building 13, Pudong International Talent Port, Lane 999, Huanke Road, Pudong New Area, Shanghai | 400 - 820 - 3556
Unit 08, 5th Floor, Building 21, No. 388 Xinping Street, Suzhou Industrial Park, Jiangsu Province | 0512 - 65107980
Room 609, Building 3, Yuhuaketing, No. 109 Ruanjian Avenue, Yuhuatai District, Nanjing, Jiangsu Province | 025 - 83531339
Room 1005, Building 1, Jiabao Kechuang Center, No. 519 2nd Avenue, Qiantang New Area, Hangzhou | 0571 - 86630817
2019 © Shanghai weichi Instrument Co., Ltd. All Rights Reserved. Design by Paiky Network.

Online Message
